Two Degrees of Freedom Genome-Wide Association Studies
Aug 15, 2020
Hello dear reader,
This is a genome-wide association study (GWAS) post, but it is a silly and frivolous one that likely doesn’t lead anywhere.
I am going to swing an idea around that I am needing to discuss regarding an analysis idea that has been plaguing me for sometime. I want to do GWAS where my fixed effect is treated as a factor, instead of continuous.
Treating the marker as a factor has several properties that I find particularly attractive, and only one negative (at least that I can think of, please comment, email, or tweet at more for futher conversation).
Pros:
1) Factors make more sense for genotypes – There is no such thing as 0.5 minor allele dosage and genotypes are not continuous, they are inherently discrete, regardless of ploidy.
2) Dominance exists – the effect of a allele substitution can differ as you include add 1 or 2 alleles. The assumption that the heterozygous value is (AA + BB) / 2 seems bad. It was stated in (CITATION) that when the observations of each marker class is unbalanced (in reality) the interpretation of the regression coeffienct is no long an allele substitution.
3) Estimating a value for each marker class enables many more comparisons – dominance and recessive behavior is better estimated when the every value of each marker class is estimated directly. This also enables direct estimation of over and under dominance. to date, I have not seen GWAS for over or under dominance despite the wealth of literature on heterosis and GWAS for heterosis. Seems silly.
Cons:
1) Treating marker classes as factors requires multiple degrees of freedom which reduces the statistical power.
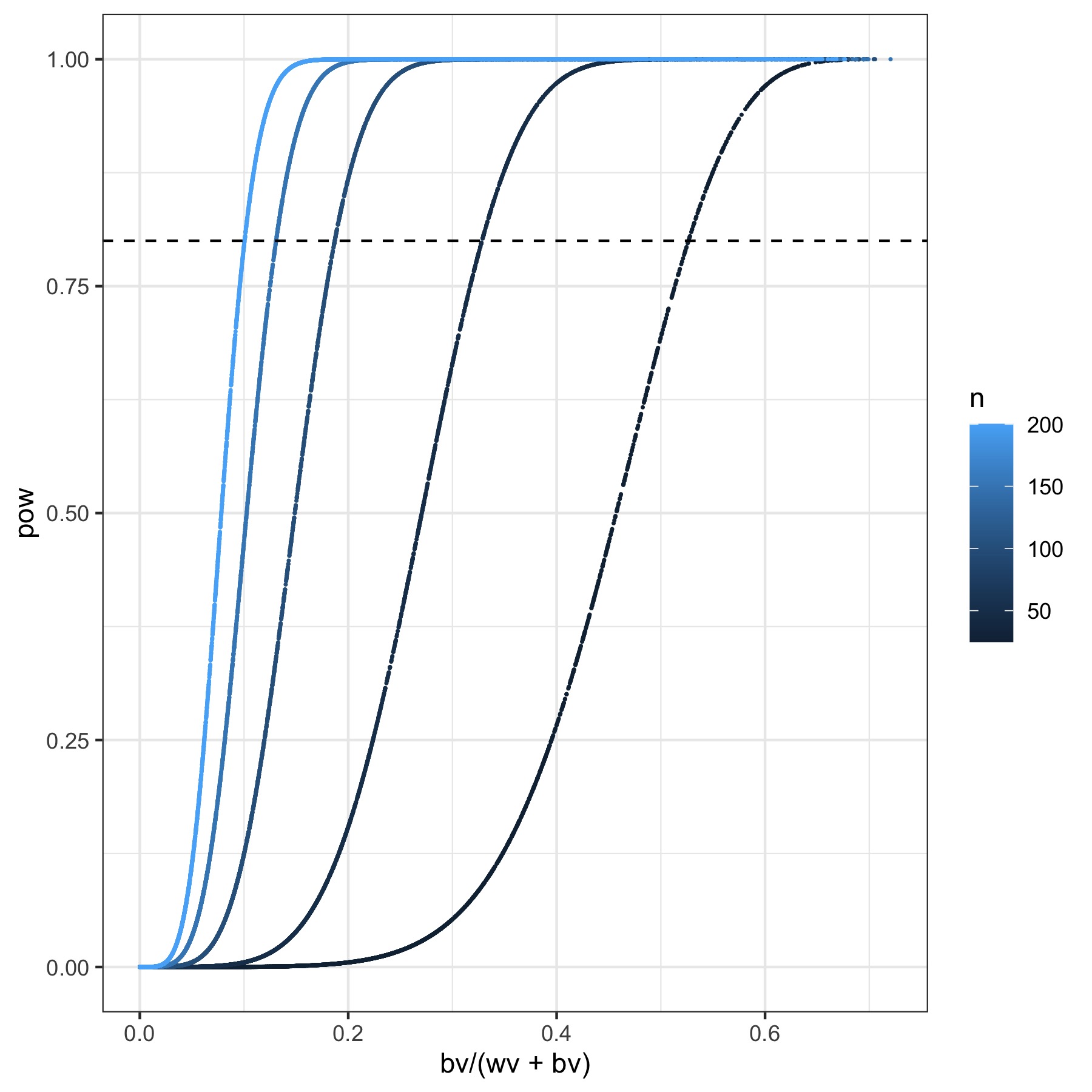
Here, I plot the statistical power associated of a one-way ANOVA with marker factor levels. The x axis is the between group variace divided by the between group plus within group variance. Colored for sample size per factor level. Obviously, this is a bit naive, but I think it starts telling an interesting story leading to interesting questions. How under/over powered are we to start implementing 2DF-GWAS?
And sure, maybe this doesn’t matter if you only work with inbred lines, but for the rest of use, why not?
Also, what is the distribution of p-values of thousands of correlated F-tests?
Best, Mitchell